Szczepionka: MRC-5 zawarta w Priorix Tetra - Całkowite sekwencjonowanie genomu

Te najnowsze analizy były możliwe dzięki aktywnemu wkładowi francuskich stowarzyszeń Liberté Informations Santé (ALIS), Ligue nationale pour la liberté des vaccinations (LNPLV) oraz Australian Association Australian Vaccination- risk Network (AVN) , za które dziękujemy.
Sekwencjonowanie nowej generacji stało się preferowanym narzędziem do pogłębionej analizy w dziedzinie biologii i nauk medycznych, zwłaszcza tych o wysokiej precyzji. Dzięki tym narzędziom możemy podejść w bardziej nowoczesny i kompleksowy sposób do wielu zastosowań, takich jak sekwencjonowanie de novo, badania metagenomiczne i epigenomiczne, sekwencjonowanie transkryptomu i ponowne sekwencjonowanie genomu.
Ten ostatni (ponowne sekwencjonowanie) jest szeroko stosowany w dziedzinie ludzkiej, zarówno do celów badawczych, jak i diagnostycznych, i składa się z NGS - sekwencjonowania nowej generacji całego pojedynczego genomu, w celu zmapowania mutacji pojedynczego nukleotydu (SNP), insercji i delecji innych lub mniej długie sekwencje, które wystąpiły w niektórych lokalizacjach genomu, oraz zmiany liczby kopii części / genomów genomu (CNV, warianty liczby kopii).
Ta procedura pomaga zrozumieć mechanizm rozwoju niektórych patologii, aby określić kierunki przyszłego leczenia klinicznego, na przykład w przypadku raka. Rzeczywiście, dzięki tej metodzie dziedzictwo genetyczne pacjenta z rakiem może być w pełni odkodowane zarówno w normalnej, jak i nowotworowej tkance, co pozwala nam zrozumieć, co dokładnie zmieniło się w genomie i, jeśli to możliwe, jak interweniować za pomocą ukierunkowanych środków.
Procedura ponownego sekwencjonowania wymaga mechanicznego rozbicia DNA osobnika na fragmenty o małych wymiarach (400-500 par zasad) i do tych fragmentów przywiązane są sztuczne części DNA o nazwie adaptery; adaptery umożliwiają związanie fragmentów ludzkiego DNA ze szklaną powierzchnią, na której wykonuje się odczyt zasad (A, C, G, T). Odczytywanie par zasad DNA odbywa się za pomocą reakcji chemicznych, a mianowicie włączenia nukleotydów, które zostały oznaczone przez cząsteczki fluorescencyjne. Tak otrzymany milion sekwencji (odczytów) jest następnie mapowany na ludzkim genomie referencyjnym za pomocą specyficznego oprogramowania i wszystkie warianty są identyfikowane poprzez porównanie analizowanego genomu z genomem referencyjnym.
Tę samą procedurę przeprowadzono na ludzkim genomie w Priorix® Tetra seria n. A71CB256A, genom należący do linii komórkowej MRC-5 (pochodzenia płodowego); prace zostały przeprowadzone przez firmę w USA, która rutynowo zajmuje się analizą ponownego sekwencjonowania genomu ludzkiego. *
* nazwa laboratorium, które wykonało analizę, zostanie uwzględniona w następnej formalnej skardze, którą złożymy u prokuratora w Rzymie, a także u włoskich i europejskich organów regulacyjnych. Stowarzyszenia, które dokonują analizy finansowanej przez Corvelvę, będą niezwłocznie informowane o tych szokujących wynikach. Nie zaprzeczamy, że czujemy się, szczególnie jako rodzice, zaniepokojeni tymi wynikami, które zgłaszamy - tak jakby to, czego do tej pory się dowiedzieliśmy, nie wystarczyło się martwić.
Wyniki
Stwierdzono, że ludzki genom referencyjny odpowiada 99,76% odczytów z DNA szczepionki, co oznacza prawie w całości. Ludzkie DNA płodu przedstawione w tej szczepionce jest pojedynczym genomem, co oznacza, że szczepionka zawiera genomowy DNA ze wszystkimi chromosomami osobnika męskiego (w rzeczywistości MRC-5 pochodzi od płodu męskiego).
Poniżej podano wyniki analizy różnych rodzajów wariantów w porównaniu z referencyjnym genomem ludzkim.
Wariant pojedynczego nukleotydu (SNP - polimorfizm pojedynczego nukleotydu) i krótkie insercje / delecje (InDels)
Warianty pojedynczych zasad DNA (SNP) to polimorfizmy, co oznacza mutacje w materiale genetycznym pojedynczego nukleotydu.
„InDels” są natomiast małymi insercjami i delecjami o długości mniejszej niż 50 pz i stanowią inną klasę wariantów genomowych w ludzkim genomie.
W ludzkim genomie szczepionki zidentyfikowano 3,6 miliona SNP (z których 98,31% jest już zgłoszonych w publicznej bazie danych dbSNP i 61,805 nowych, czyli oryginalnych w tym DNA) i około 804 000 InDels (z czego 89,42% zgłoszono już w dbSNP i 85.106 nowy).
Ilość SNP jest zgodna z tym, co opisano w literaturze na temat / w „typowym ludzkim genomie”, podczas gdy InDels daje większą ilość w porównaniu z tym, co zostało zgłoszone przez „Konsorcjum Projektu 1000 Genomes”, mianowicie 800 000 w porównaniu do 600 000 .CNV (warianty numerów kopii) i SV (warianty strukturalne)
Warianty liczby kopii (CNV) są wariantami genomowymi z powodu zmian liczby kopii stosunkowo dużych fragmentów (dłuższych niż 50 pz) między poszczególnymi genomami. Istnieją dwa rodzaje CNV: typ „zysk” (wzrost liczby kopii) i typ „utrata” (utrata kopii). 218 CNV wykryto w ludzkim genomie szczepień, z czego 82 to „zysk” (obejmujący część genomu około 6,9 miliona par zasad) i 136 CNV typu „utrata” (obejmujący część genomu około 70 milion zasad).
Jak opisano w Konsorcjum Projektu 1000 Genomes w „Globalnym źródle ludzkiej zmienności genetycznej (Nature, vol. 526, 10 października 2015)”, typowy ludzki genom zawiera od 2100 do 2500 dużych wariantów, w tym:
- 1000 dużych usunięć
- 160 wariantów w liczbie kopii (CNV)
- 10 inwersji
Które razem wpływają na 20 milionów zasad sekwencji, również biorąc pod uwagę insercje.
Jak widać w przypadku krótkich INDEL, nawet w przypadku dużych insercji i delecji genom szczepionkowy nie jest zatem zgodny z „normalnym” ludzkim genomem, ponieważ jest znacznie bardziej „przestawiony” niż genom zwykłej osoby.
Kołowe widzenie genomu (wykres cyrkowy)

Graficzna reprezentacja genomu szczepionki zwanego „polem okrężnym” (powszechnie stosowanym do przedstawienia genomu zsekwencjonowanego), jest pokazana poniżej, obok innego reprezentującego genom ponownie sekwencjonowany z DNA pobranego z krwi zdrowego osobnika - „normalny” genom :
Znaczenie koncentrycznych kół
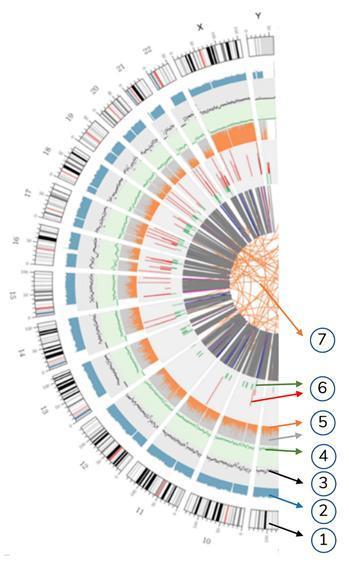
7) Najbardziej centralny pierścień reprezentuje wnioskowanie SV (warianty strukturalne) w obszarach eksonów i splicingu. TRA (pomarańczowy, translokacje), INS (zielony, wstawki), DEL (usuwanie, szary), DUP (duplikacje, różowy) i INV (inwersje, niebieski).
6) szósty okrąg reprezentuje wnioskowanie CNV (warianty numerów kopii). Kolor czerwony oznacza wzrost sekwencji DNA, a kolor zielony oznacza utratę.
5) Piąty pierścień reprezentuje udział SNP w homozygotyczności (pomarańczowy) i heterozygotyczności (szary) w układzie histogramu.
4) Czwarty pierścień (zielony) reprezentuje gęstość SNP w układzie wykresu rozrzutu.
3) Trzeci pierścień (czarny) reprezentuje gęstość INDELI w układzie wykresu rozrzutu.
2) Drugi pierścień (jasnoniebieski) reprezentuje pokrycie odczytów w układzie histogramu.
1) Zewnętrzne koło (pierwsze koło) jest liczbą chromosomów.
Można również dokonać przybliżonego porównania między DNA płodowym a DNA komórek HeLa, unieśmiertelnioną linią komórkową stosowaną również do produkcji szczepionki przeciwko polio.

Należy pamiętać, że translokacje komórek HeLa reprezentowane na wykresie woskowym przez linie jądra są odnoszone do całego genomu (stąd część kodująca i niekodująca), podczas gdy w przypadku komórek szczepionych płodowo odnoszą się tylko do genów kodujących .
Nie trzeba być naukowcem, aby zrozumieć z cyrków, po prostu na pierwszy rzut oka, że genom szczepionkowy nie jest genomem, który można określić jako „normalny”. Pomarańczowe linie splecione w centrum cyrku, niezbyt liczne w odpowiadającym mu pierścieniu „normalnego” genomu, już mają sens dla anomalii tego genomu.
Analiza wariantów genów raka
Analiza wariantów SNP, InDels, CNV, SV wybranych 560 genów, ponieważ biorą udział w różnych formach ludzkiego raka, wykazuje obecność wielu „oryginalnych” wariantów, to znaczy, że nie są one nawet obecne w publicznych bazach danych, dlatego są nieznane w literaturze. Innymi słowy, zidentyfikowano ważne modyfikacje genów, o których wiadomo, że są powiązane z różnymi postaciami nowotworów, dla wszystkich 560 zweryfikowanych genów; ponadto istnieją warianty, których konsekwencje nie są znane, ale które jednak wpływają na geny zaangażowane w indukcję ludzkiego raka.Wniosek
Ludzki genomowy DNA zawarty w szczepionce z serii Priorix. n. A71CB256A jest ewidentnie anomalny, wykazując istotne niespójności w porównaniu z typowym genomem ludzkim, tj. Genomem zdrowego człowieka. Istnieje kilka nieznanych wariantów (nie odnotowanych w publicznych bazach danych), a niektóre z nich znajdują się w genach zaangażowanych w raka. Pozornie anomalny jest również nadmiar genomu, który wykazuje zmiany w liczbie kopii (CNV) i wariantach strukturalnych (SV), takich jak translokacje, insercje, delecje, duplikacje i inwersje, z których wiele obejmuje geny.
Potencjalny wkład licznych wariantów (nieobecnych w literaturze naukowej i publicznych bazach danych) w fenotyp komórek wykorzystywanych do wzrostu wirusów szczepionkowych nie jest znany.
W pliku PDF znajdziesz wszystkie spostrzeżenia związane z implikacjami dla powitania i korespondencji z EMA.
Pobierz: CORVELVA-MRC-5-zawarty w Priorix-Tetra-Complete-sekwencjonowanie genomu




Brak komentarzy:
Prześlij komentarz